國家出手!口罩出口新規!附CE/FDA認證資格認證指南
來源: 時間:2020-04-02
商務部:4月1日起!出口醫療物資出新規


商務部昨晚發布公告:在疫情防控特殊時期,為有效支持全球抗擊疫情,保證產品質量安全、規范出口秩序,自4月1日起,出口新型冠狀病毒檢測試劑、醫用口罩、醫用防護服、呼吸機、紅外體溫計的企業向海關報關時,須提供書面或電子聲明(模版見附件1),承諾出口產品已取得我國醫療器械產品注冊證書(相關注冊信息見附件2),符合進口國(地區)的質量標準要求。海關憑藥品監督管理部門批準的醫療器械產品注冊證書驗放。上述醫療物資出口質量監管措施將視疫情發展情況動態調整。
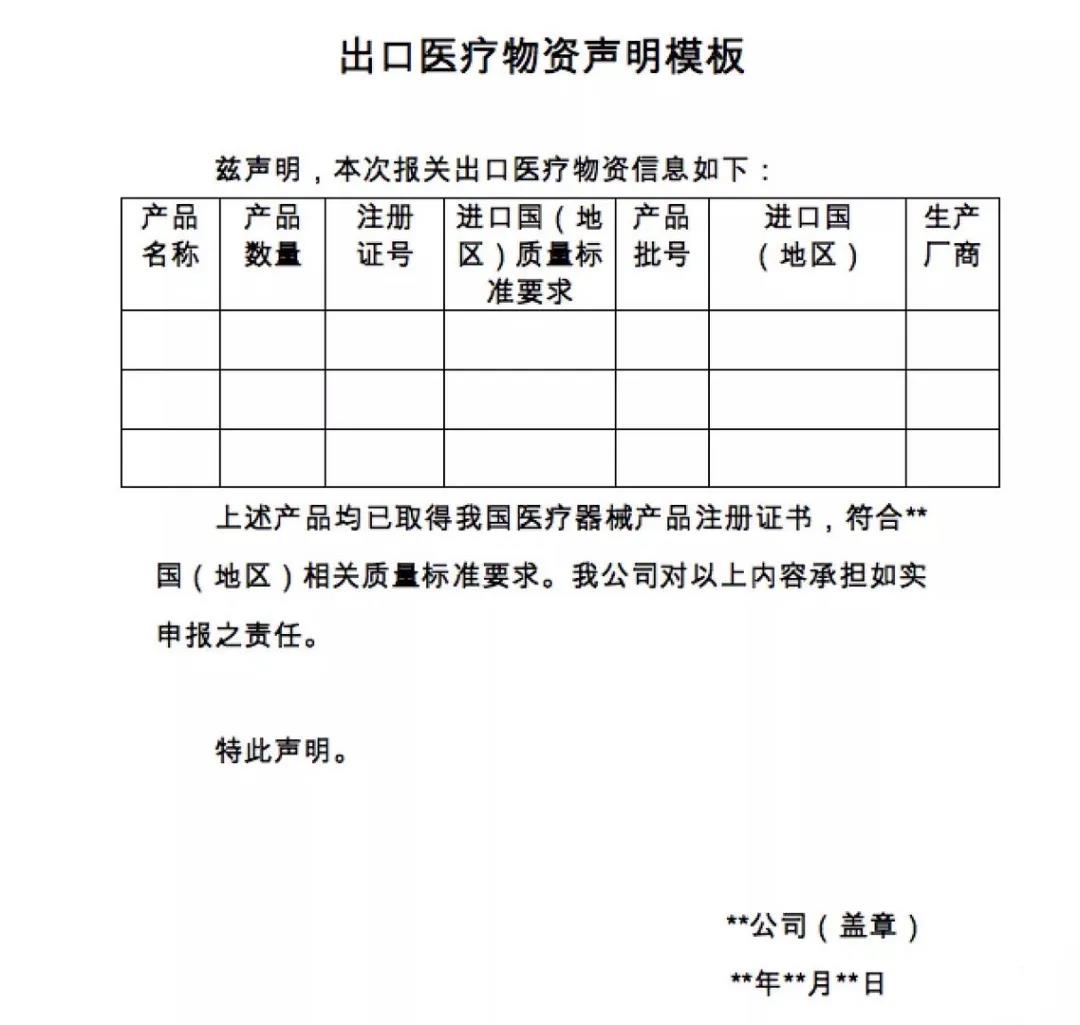
▲出口報關時新增的申明模板
根據國家商務部,海關總署和國家藥品監督管理局官方公布的已經取得我國醫療器械產品注冊證書的合格企業,全國共計2047家!
大家在詢價,采購和出口時,可以仔細從中查找你的供應商是否在三個部委公布的合規清單里面!
避免采購劣質產品,導致貨物被查被扣并最終血本無歸!
一次性使用醫用口罩(共752家)
醫用防護口罩(共150家)
醫用外科口罩(共523家)
醫用防護服(共301家)
醫用防護口罩(共23家)
呼吸機(共62家)
紅外體溫計(共236家)
歐盟CE認證資格
隨著新冠肺炎疫情的迅速蔓延,意大利、法國、德國等國紛紛淪陷,歐洲成為了疫情重災區。在防控疫情的過程中,由于醫用口罩、防護服等醫護物資的嚴重短缺,歐洲對外釋放了巨大的防護用品需求。目前,中國已經取得了新冠防疫的階段性勝利,作為世界上最大的醫用防護用品生產國,無論是傳統醫療生產企業,還是決定踏入這一陌生領域的新企業,都決定將多余的產能利用起來,將此類產品銷往歐洲市場。
出口歐盟市場,CE認證必不可少。目前市場上出現了各色各樣的醫療CE證書,讓人眼花繚亂。在各種行業微信群里,經常可以看到有人發出一張所謂的CE證書,請大家幫忙辨別真偽。為了更好地幫助到大家,下面我們就來談談具體的鑒別方法。
查詢CE證書的真偽,有多種方式,首先是最簡單粗暴的一種。大的公告機構會在自己的官網上開放查詢證書的窗口,當然,這種方式僅適用于發證機構正好提供了查詢服務的情況。而對于未開放證書查詢服務的機構,就不會奏效了。那么對于此類情況,當我們拿到一張醫療CE證書時,我們又該如何鑒別呢?
歐盟官網MDD 93/42/EEC醫療器械指令授權的機構查詢地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=13
歐盟官網MDR (EU) 2017/745醫療器械法規授權的機構查詢地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=34
歐盟官網 (EU)2016/425個人防護裝備授權的機構查詢地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=15550

自2020年5月26日起,MDR (EU) 2017/745醫療器械法規將正式取代歐盟現行的MDD醫療器械指令強制實施,同樣在歐盟官網可以查詢到,擁有MDR授權的公告機構目前只有12家。
所以,如果您手上的醫療CE證書發證機構不在以上名單范圍內,則說明它并不具備醫療產品的歐盟發證資質,更別談CE證書的發放了,那么,很遺憾地說,您拿到的這張“”CE證書“是無效的。
另外,我們也可以從醫療器械產品CE認證的流程著手去分析,完成鑒別。

美國FDA
510(k)的申請流程如下:

通常一個產品從啟動510(k),準備測試和各類文件,直至最終的審核結束,需要歷經8-10個月,而初次申請的企業,這個時間通常會更長。
除了510(k)以外,FDA要求所有的醫療器械企業都需要進行場所注冊(Establishment Registration)和產品列示(Product Listing),這一要求對于應急使用授權的產品也不例外。對于所有的海外企業,在進行場所注冊之前應先獲得鄧白氏編碼,編碼在中國由華夏鄧白氏公司代理發放,免費的代碼需要大約30天可以獲得。獲取鄧白氏編碼后,大約需要1-2周左右完成場所注冊和產品列示。
無論是510(k),場所注冊或者產品列示,FDA都不會向企業頒發任何證書,僅以美國FDA數據庫中的數據為準。也就是說,大家見過的各種有著老鷹標記的證書都是沒有任何效力的。
目前,新冠疫情在美國呈現明顯爆發的趨勢,各類醫療物資也趨于緊張。美國食品藥品監督管理局(FDA)早在今年(2020年)2月初就開始對醫療器械潛在短缺的情況進行調查,為了應對各類醫療器械的緊缺FDA發布了各類應急使用授權(Emergency Use Authorization,EUA)。目前可以申請EUA的產品主要是未獲上市的N95口罩、未獲上市的新冠病毒診斷試劑、未獲上市的酒精洗手液產品、已上市但需擴展用途的非侵入遠程監護系統、已上市和未上市的呼吸設備。
新冠檢測試劑:首當其沖的就是診斷試劑,FDA已經就該產品發布了第二版的指導原則。第一版指導原則主要針對美國國內臨床實驗室自我開發的檢驗方法的EUA申請,而第二版則納入了針對生廠商的遞交EUA申請的詳細指導。EUA的申請要包含的內容與510(k)相似,需要提交檢測試劑的描述、預期用途、性能評價報告、臨床評價方案、穩定性試驗方案、標簽標識等。由于屬于應急審批,FDA還要求企業必須提供針對患者和專業人員的明白紙(Fact Sheet)。目前海河咨詢已經完成了新冠病毒膠檢測試劑盒膠體金法的EUA申請。
(2)經NIOSH批準但已過制造商推薦的保存期限的過濾面罩口罩,供醫護人員在醫療環境中使用,以防止醫護人員由于面罩口罩短缺,而暴露于病原性生物空氣傳播顆粒中。
考慮到這樣的措施還是無法保障美國市場的口罩供應,FDA又在近期發布口罩的EUA申請,特別地,沒有獲得NIOSH批準的口罩也可以進行EUA的申請,但是必須符合:
條件1-滿足特定國家/地區性能標準,包括
澳大利亞:P2,P3
巴西:PFF2,PFF3
歐盟:FFP2,FFP3
日本:DS/DL3,DS/DL2
韓國:Special 1st
墨西哥:N100, P100, R100, N99, P99, R99, N95, P95, R95
條件2-獲得特定國家上市許可,包括:
歐盟:CE認證;
澳大利亞:ARTG注冊
加拿大:注冊證;
日本:醫療器械注冊證。
酒精洗手液產品:之前酒精洗手液產品在美國是按照非處方類藥品進行管理的,洗手液中的酒精則是按照API進行管理的。目前,FDA已經發布通知,不會對提供用于洗手液的酒精的生產商或者洗手液的生產商采取行動,即可以認為,FDA將允許用于洗手液的酒精和基于酒精的洗手液在滿足限定條件下直接在美國銷售。海河咨詢特別提醒:對于洗手液的生產商僅限定適用于藥劑師以及聯邦設施,不適用于其他商業生產商。
★ 口罩出口--海關技術性貿易措施指南 ★
一、出口通關提示
1.報關前提條件
收發貨人注冊編碼(慈善機構可為臨時編碼),需辦理無紙化通關法人卡
2.出口資質
口罩出口對生產銷售單位、境內發貨人,除滿足國內生產、市場流通資質需求外,中國海關無特殊資質要求。
3.出口申報要求
(1)商品歸類:除特殊情況外,絕大部分口罩應歸入稅號63079000。
(2)檢驗檢疫:口罩為非法檢產品,申報時檢驗檢疫項目無需填報。根據我國政府與相關國家簽訂的政府間檢驗協議,對出口伊朗等少數幾個國家的產品需按規定進行裝運前檢驗。
(3)關稅征免:如出口物資為貿易性質,征免性質申報一般征稅,征免方式申報照章征稅;如為捐贈性質,境內發貨人為貿易代理商、慈善機構等,征免性質可不填,征免方式申報全免。
(4)禁限管理:目前商務部未對口罩設置貿易管制要求,中國海關也無針對防護物資的監管證件口岸驗核要求。
(5)申報規范:按照規范申報要求填寫商品名稱、成分含量;如物資非中國生產,原產國按照實際生產國填寫。
4.出口退稅
口罩的出口退稅率為13%。
5.中美關稅排除加征
美國企業可申請排除口罩進口加征關稅,但是目前只有少數企業獲準豁免。詳見美國貿易代表辦公室網站https://ustr.gov/。
6.快速通關保障
物資出口申報如遇單窗等系統故障,可聯系現場海關采取應急方式處置,或者撥打海關12360熱線進行咨詢。
*以下內容是根據國內外相關政府機構、專業網站、新聞報道,收集整理而成,僅供參考。具體內容以相關管理部門、國外官方機構要求為準。
二、出口前準備
1.明確口罩分類
國外按照用途一般分為個人防護和醫用兩類口罩,國內出口貿易企業需具備的資質和材料如下:
(1)營業執照(經營范圍有相關經營內容)。
(2)企業生產許可證(生產企業)。
(3)產品檢驗報告(生產企業)。
(4)醫療器械注冊證(非醫用不需要)。
(5)產品說明書(跟著產品提供)、標簽(隨附產品提供)。
(6)產品批次/號(外包裝)。
(7)產品質量安全書或合格證(跟著產品提供)。
(8)產品樣品圖片及外包裝圖片。
(9)貿易公司須取得海關收發貨人注冊備案。
2.國內出口口罩生產企業資質證明
生產個人防護或者工業用非醫療器械管理的普通口罩,有進出口權的企業,可自行直接出口。
生產屬于醫療器械管理的口罩用于出口,中國海關不需要企業提供相關資質證明文件,但一般進口國會要求生產企業提供產品三證,以證明該進口的商品在中國已合法上市,具體如下:
(1)營業執照(經營范圍包含有醫療器械相關,非醫療級別的物品不需要)。
(2)醫療器械產品備案證或者注冊證。
(3)廠家檢測報告。
生產企業有進出口權,可以自行出口,如沒有進出口權,可以通過外貿代理進行出口銷售。
3.內貿企業做出口需要取得的基本資質
(1)向市場監管部門取得營業執照,增加經營范圍“貨物進出口、技術進出口、代理進出口”。
(2)向商務部門取得進出口權,可直接在商務部業務系統統一平臺(http://iecms.mofcom.gov.cn/)申請,網上提交材料。
(3)向外匯管理局申請取得開設外匯賬戶許可。
(4)辦理進出口貨物收發貨人海關注冊登記。
三、各國口罩準入條件(產品準入條件)
1.美國
必要資料:提單,箱單,發票。
個人防護口罩:必須取得美國 NIOSH檢測認證,即National Institute for Occupational Safety and Health美國國家職業安全衛生研究所認證。
醫用口罩:須取得美國FDA注冊許可。
2.歐盟
必要資料:提單,箱單,發票。
個人防護口罩:個人防護口罩的歐盟標準是EN149,按照標準將口罩分為FFP1/FFP2和FFP3三個類別。所有出口歐盟的口罩必須獲得CE認證證書。CE認證是歐盟實行的強制性產品安全認證制度,目的是為了保障歐盟國家人民的生命財產安全。
醫用口罩:醫用口罩對應的歐盟標準是EN14683。
產品在歐盟銷售需要出具歐盟自由銷售證書 Free Sale Certificate,有了CE標志并進行了相關指令中要求的歐盟注冊后,中國的制造商出口歐盟不需要自由銷售證書。
3.日本
必要資料:提單,箱單,發票,日本國外的制造商必須向PMDA注冊制造商信息。
口罩包裝要求:包裝上印有ウィルスカット(中文翻譯:病毒攔截)99%的字樣
PFE:0.1um微粒子顆粒過濾效率
BFE:細菌過濾率
VFE:病毒過濾率
口罩品質標準:
(1)醫用防護口罩:符合中國GB 19083-2010 強制性標準,過濾效率≥95%(使用非油性顆粒物測試)。
(2) N95口罩:美國NIOSH認證,非油性顆粒物過濾效率≥95%。
(3) KN95口罩:符合中國GB 2626 強制性標準,非油性顆粒物過濾效率≥95%。
4.韓國
必要資料:提單,箱單,發票,韓國進口商營業執照。
個人防護口罩標準:KF (Korean filter) 系列分為KF80、KF94、KF99
執行標準規范:MFDS Notice No. 2015-69
韓國醫療器械準入的法規門檻,基本分類為I、II、III、IV類,持證為韓國公司(License holder),韓國收貨人需要到韓國藥監局Korea Pharmaceutical Traders Association. 提前備案進口資質(沒有不行)網址:www.kpta.or.kr。
5.澳大利亞
必要資料:提單,箱單,發票。
須通過澳洲的TGA注冊,符合標準規范:AS/NZS 1716:2012,此規范是澳大利亞和新西蘭的呼吸保護裝置標準。
TGA 是Therapeutic Goods Administration的簡寫,全稱是治療商品管理局,它是澳大利亞的治療商品(包括藥物、醫療器械、基因科技和血液制品)的監督機構。澳大利亞對醫療器械分為I類,Is and Im, IIa, IIb, III類,產品的分類幾乎和歐盟分類一致,如果產品已經獲得CE標志,則產品類別可以按照CE分類。
四、各國注冊、認證簡要辦理流程
1.美國NIOSH認證
需按照NIOSH的指南實施,企業需寄送樣品至NIOSH實驗室實施測試,同時提交技術性資料(包括質量體系部分資料)至NIOSH文審,只有文審和測試都通過,NIOSH才核發批文。NIOSH將其認證的防顆粒物口罩分為9類,具體的測試則由NIOSH下屬的NPPTL (National Personal Protective Technology Laboratory)實驗室操作。主要測試指標包括呼氣阻力測試、呼氣閥泄漏測試、吸氣阻力測試、過濾效率測試。
2.美國FDA注冊

3.歐盟CE注冊

4.日本PMDA注冊

(1)準備階段。確定產品分類(I,II特殊控制,II類控制,III,IV)和產品JMDN編碼,選擇MAH(日本持證方);
(2)制造商向PMDA注冊工廠;
(3)II類特殊控制產品向授權認證機構PCB申請QMS工廠審核,其他II類產品和III類IV類產品向PMDA申請QMS工廠審核,并獲得QMS證書;
(4)申請Pre-Market Apporval證書,II類特殊控制由PCB發證,其他II類產品和III類IV類產品控制由MHLW(厚生勞動省)發證;
(5) 支付申請費用;
(6) 注冊文件整改,注冊批準;
(7)所有類別產品均需要MAH向RBHW(厚生省地區機構)進行進口通報注冊后才能進口銷售。
5.韓國KFDA注冊
韓國衛生福利部(MinistryofHealthandWelfare,MHW),簡稱衛生部,主要負責管食品、藥品、化妝品和醫療器械的管理,是最主要的衛生保健部門。依照《醫療器械法》,韓國衛生福利部下屬的食品藥品安全部負責對醫療器械的監管工作。KFDA注冊流程為:
(1)確定產品分類(I,II,III,IV),選擇KLH(韓國持證方);
(2)II類產品需申請KGMP證書和接受現場審核,II類產品一般是授權的第三方審核員,并獲得KGMP證書;
(3)II類產品需要送樣品到韓國MFDS授權的實驗室進行韓國標準的測試;
(4)由KLH向MFDS(韓國食品藥品安全部)提交技術文件(檢測報告,KGMP證書等),進行注冊審批;
(5)支付申請費用;
(6)注冊文件整改,注冊批準;
(7)指定韓國代理商和經銷商,產品銷售。
6.澳大利亞TGA注冊
依據Australian Therapeutic Goods (Medical Devices) Regulations 2002,澳大利亞對醫療器械分為I類,Is and Im, IIa, IIb, III類,產品的分類幾乎和歐盟分類一致,如果產品已經獲得CE標志,則產品類別可以按照CE分類。如果已經獲得歐盟公告機構(Notified Body)簽發的CE證書,是可以被TGA認可的,并可以作為滿足澳大利亞安全法規的重要注冊資料。

五、各國口罩技術標準對比(供生產企業參考)



六、各國口罩技術標準(供生產企業參考)
|
序號 |
標準號 |
標準名稱 |
狀態 |
發布時間 |
|
國際 |
ISO 22609:2004 |
傳染試劑防護服.醫療面罩.防人造血滲透的試驗方法(固定容積,水平注射) |
現行 |
2004/12/3 |
|
歐盟 |
EN 136-1998 |
呼吸保護裝置.全面罩.要求,試驗,標記。 |
現行 |
1998/1/1 |
|
EN 140-1998+AC-1999 |
呼吸保護裝置.半面罩和四分之一面罩.要求,試驗和標記。 |
現行 |
1998/9/1 |
|
|
EN 143-2000 |
呼吸防護裝置.微粒過濾器.要求,試驗,標記。 |
現行 |
2000/2/1 |
|
|
EN 149-2001 |
呼吸防護裝置.顆粒防護用過濾半面罩.要求,檢驗和標記。 |
現行 |
2001/4/1 |
|
|
EN 529-2005 |
呼吸保護裝置.選擇,使用,保養和維修的建議。 |
現行 |
2005 |
|
|
EN 12942-1998 |
呼吸保護器.帶全面罩,半面罩和四分之一面罩的鼓風過濾裝置.要求,檢驗,標識。 |
現行 |
1998 |
|
|
EN 14387-2004+A1-2008 |
呼吸保護裝置.氣體過濾器和組合過濾器.要求、測試、標記。 |
現行 |
2004/1/1 |
|
|
EN 14683-2019 |
醫用口罩 要求和試驗方法。 |
現行 |
2019/3/1 |
|
|
美國 |
ASTM F1862/F1862M-2017 |
醫用口罩抗人工合成血滲透的標準試驗方法(已知速度下固定體積的水平投影)。 |
現行 |
2017 |
|
ASTM F2100-2019 |
醫用口罩材料性能標準規范。 |
現行 |
2019 |
|
|
ASTM F2101-2019 |
用金黃色葡萄球菌生物氣溶膠評價醫用口罩材料的細菌過濾效率(BFE)的標準試驗方法。 |
現行 |
2019 |
|
|
ASTM F2299/F2299M-2003(2017) |
用膠乳球測定醫用面具材料粒子滲透性初始效率的標準試驗方法。 |
現行 |
2003 |
|
|
澳大利亞 |
AS/NZS 1715:2009 |
呼吸保護設備的選擇,使用和維護。 |
現行 |
2009/2/6 |
|
AS/NZS 1716:2012 |
呼吸保護裝置。 |
現行 |
2012/2/13 |
|
|
日本 |
JIS T 8062:2010 |
預防傳染性病原體的防護服.面罩.防止人造血漿滲透的試驗方法(確定容量,平行注射)。 |
現行 |
2010/5/25 |
|
JIS T 8159:2006 |
呼吸防護設備的選擇、使用和維護指南。 |
現行 |
2006/4/25 |
|
|
JIS T 8159:2006 |
呼吸保護裝置泄漏率試驗方法。 |
現行 |
2006/2/20 |
|
|
韓國 |
KS M 6673-2008 |
防塵口罩 |
現行 |
2008/2/22 |
|
KS K ISO 22609-2018 |
傳染試劑防護服.醫療面罩.防人造血滲透的試驗方法(固定容積、水平噴射)。 |
現行 |
2018/11/14 |
*以上技術標準如有動態調整,以相關標準管理機構官方發布為準。
Copyright @ 2017 山東省信達雅國際商貿有限公司版權所有 . 魯ICP備11006413號-1
電話: 0531-86080340 | 86086106 | 86086107
地址:濟南市泉城路院前街珍珠泉大院(省人大大院)9號樓5樓













